






Опиатная наркоманияКраткое описание:
Опиатная наркомания является важной медицинской и социальной проблемой. Фундаментальные исследования многих лабораторий свидетельствуют о существенном воздействии опиатов на трансдукторные системы и экспрессию генов. Такие результаты позволяют предполагать, что изменения в системах вторичных мессенджеров и в экспрессии генома, вызванные морфином и другими мю-опиатными агонистами, могут быть вовлечены в формирование толерантности, пристрастия и абстинентного синдрома. В данной работе обсуждаются некоторые из указанных проблем.
Нейрохимия опиатной наркомании
А.И.Головко*, Д.А.Коноплин, Ю.А.Некрасов, О.И.Романенко, М.А.Стрельский Региональный медицинский лечебно-диагностический центр "Бехтерев", 196083 Санкт-Петербург, ул. Корабельная, 6. Тел. (812) 183-9357, факс (812) 328-1324, E-mail: psycho@narcom.ru Ключевые слова: опиаты, наркомания, нейрохимия, трансдукторные системы, геном, экспрессия, толерантность, пристрастие, абстинентный синдром. ВведениеВ патогенезе опиатной наркомании значительную роль играют нарушения взаимодействия нейромедиаторных систем [1, 2, 3, 4]. Это касается практически всех изученных к настоящему времени нейротрансмиттеров: опиоидов, дофамина, норадреналина, серотонина, глутаминовой кислоты, ГАМК, глицина и т.д. Сдвиги в системах нейропередачи имеют непосредственное отношение к формированию феномена пристрастия, абстинентного синдрома, толерантности. В практическом плане существующая ныне фармакотерапия опиатной наркомании во многом направлена на коррекцию нарушенных связей между нейромедиаторными системами [5, 6, 7, 8, 9, 10]. Основными средствами, имеющимися в арсенале современной наркологии, по-прежнему считаются анальгетики, нейролептики, транквилизаторы, ноотропы, блокаторы кальциевых каналов, адренотропные средства, антидепрессанты [1, 3, 5, 11]. Считается, что аддиктивный потенциал наркотических средств реализуется не только на уровне синаптической передачи, но включает также изменения систем вторичных месенджеров и даже генома [12]. С этой точки зрения вполне понятны попытки исследователей использовать не только синаптотропные фармакологические препараты, но и средства, модулирующие системы вторичных посредников. В данном контексте показательны успехи отечественных наркологов, предложивших использовать в фармакотерапии опиатной наркомании блокаторы кальциевых каналов [12]. Итак, нарушения взаимодействия нейромедиаторных систем могут считаться начальным звеном патогенеза опиатной наркомании. Они же являются мишенью фармакотерапии при лечении абстинентного синдрома и в период поддержания ремиссии. В данном обзоре предпринята попытка обобщить некоторые нейрохимические аспекты опиатной наркомании: функционирование опиоидных нейромедиаторных систем, нейрохимические основы толерантности и зависимости, состояние систем вторичных месенджеров и генома при наркомании. I. Опиоидные рецепторыОпиоиды (эндорфины, энкефалины и динорфины) относятся к числу пептидных нейротрансмиттеров. Важным элементом опиоидной нейромедиаторной системы являются соответствующие рецепторы. Предположения о наличии опиоидных рецепторов (ОР) высказаны еще в середине 50-х гг. 20-го столетия. В 70-х годах выделены эндогенные лиганды ОР эндорфины, энкефалины и динорфины. В первой половине 90-х гг. осуществлено клонирование рецепторов [13, 14, 15, 16]. Первоначальное обозначение классов ОР мю, каппа и дельта в основном определялось названием определенного лиганда. Так, мю-рецепторы обозначены в соответствии с высоким сродством к агонисту морфину, каппа-рецепторы чувствительны к кетоциклазоцину. Подобные обозначения не отражали сродства рецепторов к их эндогенным лигандам. Как результат - наличие нескольких классификаций ОР (табл.1). Опиоидные рецепторы относятся к семейству метаботропных, т. е. передача информации внутрь нейрона после связывания с агонистом осуществляется путем модуляции различных систем вторичных месенджеров, в первую очередь аденилатциклазной [18, 19]. Есть сведения о наличии мест специфического связывания опиоидов в пределах кальциевых каналов [19, 20], а также об их способности влиять на обмен калия и натрия [19, 20, 21].
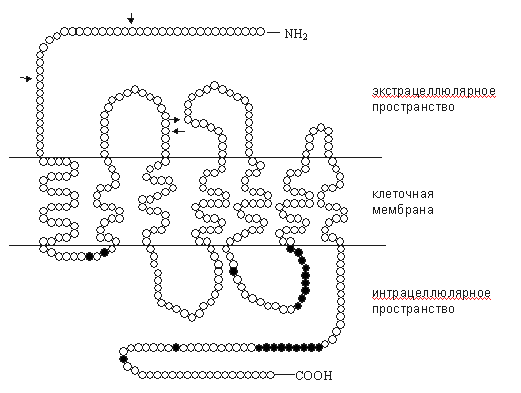
Рис. 1. Схема опиоидного рецептора [21, 27], с дополнениями. Полипептидная цепь опиоидных рецепторов семь раз пронизывает нейрональную мембрану. Соответственно, трансмембранные участки обозначают ТМ-1 - ТМ-7. Трансмембранные домены ассоциированы с гуаниннуклеотидсвязывающими белками - G-белками. NH2-терминаль, экстрацеллюлярные петли и верхушка ТМ-4 являются участками связывания агонистов и антагонистов. Однако для каждого рецептора участки рецептирования лигандов различны: в дельта-рецепторах это третья экстрацеллюлярная петля, в мю-рецепторах - первая и третья экстрацеллюлярные петли, в каппа-рецепторах - 2-я экстрацеллюлярная петля и верхушка ТМ-4 [19, 21, 22]. Пока остается открытым вопрос о ключевых аминокислотных остатках, участвующих в рецептировании. Решение этой проблемы обеспечивает дальнейший прогресс в разработке новых фармакологических средств. Основным направлением таких исследований остается клонирование мутантных форм опиоидных Таблица 1
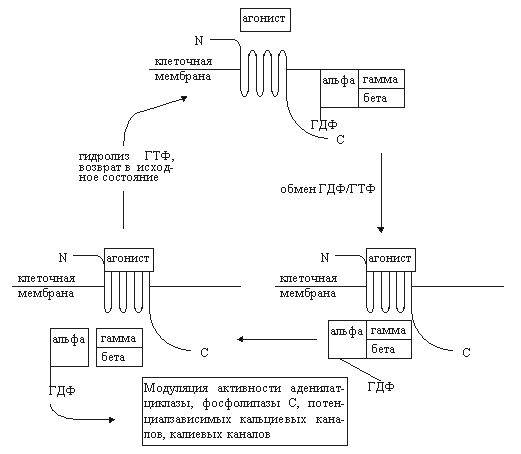
Примечания: * - номер класса отражает динамику исследований по клонированию рецепторов: ОР1 клонированы раньше, чем ОР2 и ОР3; в современной научной литературе опиоидные рецепторы наиболее часто обозначают первоначальными аббревиатурами: мю, дельта и каппа; самостоятельность других классов опиоидных рецепторов (эпсилон, кси и ламбда) к настоящему времени признается не всеми исследователями рецепторов[22, 23, 24, 25, 26]. C-терминаль и интрацеллюлярные петли имеют несколько участков для фосфорилирования с помощью протеинкиназ, регулируемых циклическим аденозинмонофосфатом (цАМФ) и протеинкиназой С (рис. 1). Наиболее изученными путями трансдукции интрацеллюлярного сигнала с участием опиоидных рецепторов являются модуляция активности аденилатциклазы, фосфолипазы С, потенциалзависимых кальциевых каналов и калиевых каналов. Все названные пути передачи информации предполагают участие G-белков (рис. 2).
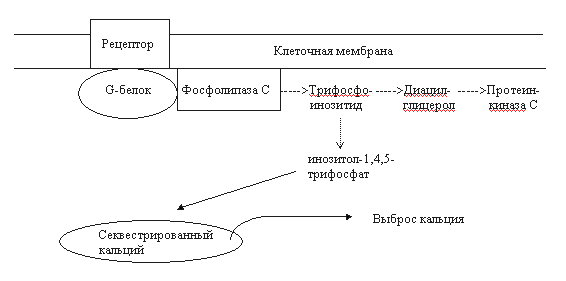
Рис. 2. Схема взаимодействия G-белка с опиоидным рецептором (цикл G-белка) [18]. Объяснения в тексте. G-белок - гетеромерный белок, ассоциированный с мембраной. Он включает альфа-, бета- и гамма-субъединицы. В покое все три субъединицы связаны между собой, а альфа-субъединица взаимодействует с гуанозиндифосфатом (ГДФ). После рецептирования агониста облегчается связывание опиоидного рецептора с G-белком. Далее ГДФ заменяется на гуанозинтрифосфат (ГТФ) в альфа-субъединице, а весь комплекс G-белка диссоциирует на два фрагмента: "альфа" и "бета-гамма". Свободные субъединичные комплексы способны взаимодействовать с эффекторами аденилатциклазой, фосфолипазой С, калиевыми и кальциевыми каналами. альфа-Субъединица, обладающая внутренней ГТФ-азной активностью, гидролизует ГТФ до ГДФ после взаимодействия с эффектором. При этом каталитическая активность субъединицы теряется, она диссоциирует из комплекса с эффектором. В последующем происходит реассоциация всех субъединиц, и система возвращается в исходное состояние [18]. Гуаниннуклеотидсвязывающие белки отличаются гетерогенностью, что, в свою очередь, определяется множественностью изоформ входящих в их состав субъединиц. Например, идентифицированы 16 типов альфа-субъединицы, 7 – бета- и 5 - гамма-субъединицы. Эффекторы, с которыми взаимодействуют G-белки, также неоднородны. Так, существует не менее десяти разновидностей аденилатциклазы [18]. Все это предопределяет множественность путей трансдукции, медиируемых G-белками. Вторым по значимости путем передачи внутриклеточного сигнала при активации опиоидных рецепторов следует считать фосфатидилинозитидный или инозитолфосфатный цикл. Фосфоинозитиды являются важнейшими компонентами нейрональной мембраны. Стимуляция опиоидных рецепторов приводит к активации фосфолипазы С через гуаниннуклеотидсвязывающий белок. Фосфолипаза С индуцирует гидролиз фосфатидил-4,5-дифосфата (трифосфоинозитид). При этом образуется 2 важнейших продукта - диацилглицерол и инозитол-1,4,5-трифосфат (рис. 3). Оба вещества считаются вторичными мессенджерами. Диацилглицерол является мощным эндогенным активатором протеинкиназы С. Этот фермент, также как и циклонуклеотидзависимые протеинкиназы, фосфорилирует регуляторные белки и тем самым изменяет физиологическую активность клетки. Инозитол-1,4,5-трифосфат стимулирует выброс кальция из внутриклеточных депо. Кальций, являющийся вторичным мессенджером, оказывает влияние на активность клетки через систему кальций-кальмодулинзависимых протеинкиназ. Краткая характеристика опиоидных рецепторов.Рецепторы типа ОР1 (дельта-рецепторы). Доказано существование минимум двух подтипов ОР1: дельта1- и дельта2-рецепторы (ОР1А и ОР1В). Эндогенными лигандами ОР1 являются лей- и метэнкефалины, предшественником которых является проэнкефалин А [17, 19, 28, 29]. Синтетические лиганды этих рецепторов - BW373U86 и SNC80 (агонисты), а также ICI154.129, ICI174.864, калтриндол, TIPP, TIPP(пси) (антагонисты). Плотность ОР1 в головном мозге млекопитающих значительно ниже в сравнении опиатными рецепторами других типов. Их преимущественная локализация - обонятельные луковицы, стриатум, неокортекс и прилежащее ядро. Концентрация ОР1 в стволе, таламусе и гипоталамусе существенно ниже [17]. ОР1-рецепторы участвуют в регуляции многих физиологических процессов: болевой чувствительности (в том числе и на спинальном уровне), когнитивных функций, настроения, зрения, дыхания, двигательной активности. Показано вовлечение ОР1-рецепторов в ингибирование эвакуаторной функции кишечника. Рецепторы типа ОР2 (каппа-рецепторы). Существует не менее трех подтипов каппа-рецепторов: каппа1-, каппа2- и каппа3-рецепторы. Наиболее изученными считаются каппа1-рецепторы. Возможно, имеется лишь один сайт этих рецепторов, меняющий свою аффинность в зависимости от особенностей взаимодействия с G-белками. Предшественником эндогенных агонистов каппа-рецепторов динорфинов А и Б является продинорфин. К агонистам относятся также кетоциклазоцин, этилкетоциклазоцин, бремазоцин, бензодиазепин тифлуадом. Среди антагонистов данных рецепторов наибольшее сродство проявляет норбиналторфимин. ОР2-рецепторы вовлечены в регуляцию нейроэндокринной секреции, диуреза, ноцицепции, потребления пищи. Они обнаружены также на иммунокомпетентных клетках [30, 31]. Следует отметить, что в фармакологическом плане наблюдаются реципрокные отношения между мю- и каппа-опиоидными рецепторами [32, 33]. мю-опиоидные рецепторы (ОР3-рецепторы). Наиболее изученный тип. Как и в случае каппа-рецепторов, подразделение на 2 подтипа не может считаться вполне доказанным, так как это может быть одна популяция рецепторов, ассоциированная с различными G-белками [17]. Эндорфины, эндогенные агонисты мю-рецепторов, образуются путем протеолитической деградации предшественника проопиомеланокортина. В ткани мозга обнаружен также эндогенный морфин, являющийся частичным агонистом мю-рецепторов. Кроме морфина, агонистами этих рецепторов являются фентанил, суфентанил, оментанил, аналог мет-энкефалина FK 33.824, а также пептиды DAMGO, DAGO, DAGOL. К антагонистам относят налоксон, налтрексон, налоксазон, налоксоназин и др. Плотность мю-рецепторов в зависимости от структуры головного мозга выглядит следующим образом: стриатум > неокортекс > таламус > прилежащее ядро > гиппокамп > миндалина. Выявляются они в задних рогах спинного мозга. Менее богаты мю-рецепторами околопроводное серое вещество и ядра шва. Очень низка их плотность в гипоталамусе. Большую группу составляют периферические мю-рецепторы.
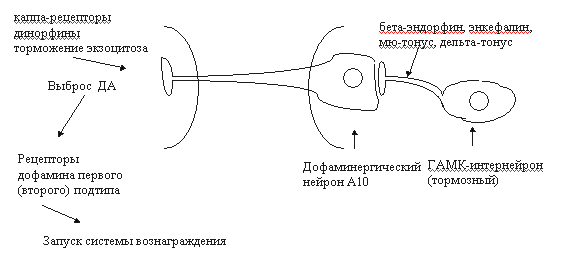
Рис. 3. Упрощенная схема фосфатидилинозитидного цикла [18]. Среди функций, регулируемых ОР3-рецепторами, следует отметить ноцицепцию, дыхание, память, обучение, секрецию нейрогормонов, сократительную активность кишечника и другие (см. обзор [17]). В конце 1995 г. сформировалось представление о наличии особой пептидергической нейромедиаторной системы, передача в которой осуществляется с участием нейропептида ноцицептина или орфанина FQ. По структуре и функциям орфановые рецепторы очень близки к опиоидным, поэтому их нередко называют ORL-1 рецепторами (opioid receptor like-1) [34, 35, 36]. Идентичность по аминокислотам у рецепторов ORL-1 в сравнении с опиоидными рецепторами достигает 63-65%. Наибольшая гомология - с каппа опиоидными рецепторами [35]. Орфановый рецептор мыши состоит из 367 аминокислотных остатков, а человека - из 370. В ЦНС млекопитающих ORL-1-рецепторы расположены во многих структурах: в миндалине, гиппокампе, ножке эпифиза, перегородке, гипоталамусе, стволе головного мозга (голубое пятно, парабрахиальные ядра, дорсальные ядра шва), в стриатуме, мозжечке, в дорсальных и вентральных рогах спинного мозга. Обнаружены ORL-1-рецепторы и в периферических тканях, в том числе на клетках иммунной системы [35]. Эндогенный лиганд ORL-1-рецепторов ноцицептин/орфанин FG состоит из 17 аминокислотных остатков, что соответствует длине лиганда каппа-рецепторов динорфина А. Ноцицептин значительно короче бета-эндорфина, но длиннее мет- и лей-энкефалинов. Передача сигнала рецепторами ORL-1 осуществляется с участием сопряженных G-белков посредством модуляции активности аденилатциклазы, тока К+ внутрь клетки и потенциалзависимых кальциевых каналов[34, 35]. Особо чувствительны к возбуждению ORL-1-рецепторов кальциевые каналы N-типа (регулируют экзоцитоз нейротрансмиттеров). Доказано участие рецепторов ORL-1 в регуляции процессов ноцицепции (гиперальгезия), памяти и обучения, внимания, эмоций, локомоции, нейроэндокринной секреции, зрения, вкуса, потребления пищи, сокращения гладкой мускулатуры, иммуногенеза [35]. Появляются сообщения об участии ноцицептина и его рецепторов в формировании толерантности к опиатам [37]. Как известно, одним из основных компонентов формирования пристрастия является активация церебральной системы вознаграждения (reward system). Центральное звено этой системы - дофаминергические нейроны А10 вентральной области покрышки (ventral tegmental area) и проекции этих нейронов в прилежащее ядро (nucleus accumbens) и в префронтальную кору [3, 38]. Тоническая активация системы вознаграждения медиируется высвобождающимся в прилежащем ядре дофамином через D1- и, возможно, через D2-рецепторы (рис. 4). К нейроанатомическим субстратам системы награды относят также голубое пятно, миндалину, околопроводное серое вещество, латеральный гипоталамус, шов, бледный шар [3, 38, 39, 40, 41, 42]. В регуляции функциональной активности ДА-ергической мезолимбической системы вознаграждения принимают участие опиоидные рецепторы всех трех типов. мю- и дельта-Опиоиды активируют ДА-ергические нейроны А10 вентральной области покрышки опосредованно - посредством блокирования тормозных ГАМК-интернейронов (рис. 4). При этом усиливается базальная секреция дофамина в nucleus accumbens, и активируется система вознаграждения [38, 39, 40]. Каппа-рецепторы тормозят экзоцитоз дофамина в прилежащем ядре (пресинаптическое торможение). Подавление выброса дофамина в nucleus accumbens сопровождается развитием синдрома отмены (дисфория, тревожность и др.). Такие эффекты вызывают каппа-агонисты. Активация ДА-ергической мезолимбической системы награды связана с мю- и дельта1-опиоидными рецепторами, а дельта2-агогнисты могут инициировать эффекты вознаграждения и без участия дофаминовой нейротрансмиссии [43]. Считается, что дофаминергический мезолимбический путь - общая мишень для веществ, влияющих на мотивации (аддиктивные или наркогенные агенты) [12, 35, 39, 40]. II. Системы вторичных мессенджеров при опиатной наркомании. Нейрохимические проблемы толерантности и абстинентного синдромаМодуляция систем вторичных передатчиков при хронической опиатной интоксикации имеет прямое отношение к формированию толерантности и абстинентного синдрома. Наиболее изучены изменения системы цАМФ, фосфоинозитидного каскада, кальциевого гомеостаза и обмена оксида азота (NO) [12, 18, 44, 45]. Негативные отношения между опиатами/опиоидами и аденилатциклазной системой первоначально показаны на клеточных культурах (см. обзоры [18, 19]). Как оказалось, эти процессы требуют присутствия Na+ и гуанозинтрифосфата (ГТФ). Торможение аденилатциклазы опиоидными рецепторами осуществляется через G-белки. Длительное воздействие опиатами сопровождается изменением активности как самой аденилатциклазы, так и сдвигами сопряженных систем: G-белков, протеинкиназ. Функциональные нарушения различных компонентов циклазного каскада носят отчетливый региональный характер (больше в тех структурах, которые вовлечены в системы вознаграждения), периода и способа наркотизации, химической структуры аддиктивного агента и других факторов. Р.Тервиллиджер и др. [45] показали, что наркотизация крыс морфином сопровождалась достоверным повышением активности аденилатциклазы и цАМФ-зависимой протеинкиназы в прилежащем ядре, голубом пятне, миндалине и таламусе. В гиппокампе, мозжечке, черной субстанции, вентральной области покрышки и в околопроводном сером веществе изучаемые показатели оставались в пределах нормы. Значительно изменялись уровни G-белков. Так, в прилежащем ядре отмечено понижение содержания Giальфа, а уровни белков Gоальфа, Gsальфа и Gбета не изменялись. В миндалине возрастали концентрации Giальфа- и Gоальфа-белков, в то время как содержание Gsальфа и Gбета было стабильным. В таламусе изучаемые показатели не отличались от контрольных. Авторы полагают, что выявленные сдвиги могут быть вовлечены в механизмы наркотической толерантности, зависимости и абстинентного синдрома. Это подтверждается сходными изменениями аденилатциклазной системы при наркотизации животных другим аддиктивным веществом - кокаином. Препараты, не обладающие наркогенным эффектом (галоперидол, имипримин, флуоксетин, клонидин) в аналогичных условиях эксперимента не влияли на изучаемые показатели. Героин вызывал сходные изменения некоторых элементов системы цАМФ. Так, в прилежащем ядре крыс, получавших этот наркотик, выявлялось повышение активности растворимой и мембраносвязанной протеинкиназы. Напротив, иммунореактивность, относящаяся к гуаниннуклеотидсвязывающему белку Giальфа, достоверно понижалась. В черной субстанции, стриатуме изучаемые показатели не отличались от контрольных значений [41]. Одной из причин дезинтеграции аденилатциклазной системы при развитии толерантности может быть нарушение взаимодействия опиоидных рецепторов с соответствующими G-белками. Так, если нативные дельта-опиоидные рецепторы ассоциированы Giальфа1-, Giальфа3- и Gоальфа-белками, то в условиях воздействия агониста-наркотика они связываются с G-белком типа Giальфа2 [19]. Сходные результаты получены и в опытах на других опиоидных рецепторах. Особое значение в развитии толерантности и зависимости придается способности рецепторов взаимодействовать с гуаниннуклеотидсвязывающим белком GS. Показано, что модификация активности определенных G-белков оказывает влияние на функциональное состояние систем вознаграждения. Например, локальное введение коклюшного токсина (ингибитор гуаниннуклеотидсвязывающих белков Giальфа и Gоальфа) в прилежащее ядро крыс сопровождалось активацией поведения самовведения героина и кокаина [42]. Как известно, система циклического аденозинмонофосфата регулирует биохимические процессы в клетке посредством фосфорилирования белков-мишеней [46]. Можно предположить, что нарушения в циклазной системе при наркотизации могут быть причиной последующих изменений на уровне фосфорилирования. Конечный результат - дизрегуляторное влияние на пластические процессы, энергообеспечение клетки, ионные каналы, стабильность биологических мембран, функциональное состояние генома. Дезинтеграция многочисленных биохимических процессов может считаться основой формирования пристрастия, толерантности и абстинентного синдром. Если к этому добавить развивающиеся параллельно сдвиги в каскадах других вторичных мессенджеров (кальция, трифосфоинозитида, диацилглицерола, циклического гуанозинмонофосфата, оксида азота), то сложность патогенеза наркозависимостей неизмеримо возрастает.
Рис. 4. Схема взаимодействия опиоидных и дофаминергических систем вентральной области покрышки и прилежащего ядра [29]. Наиболее длительным нарушением считается дезадаптационная экспрессия определенных участков генома в ответ на поступление в организм агента, обладающего аддиктивным потенциалом. Такие изменения, даже после однократной инъекции, могут сохраняться несколько лет, а возможно - и всю жизнь [12]. Традиционно в патогенезе опиатной наркомании важное место отводили изменениям опиоидных рецепторов (десенсистизация, down-regulation), дезинтеграции связей "опиоидные рецепторы - системы вторичных посредников", нарушениям дофаминергических и катехоламинергических нейромедиаторных систем [18, 43, 44, 47, 48]. В последние годы внимание исследователей привлекают и другие нейрохимические системы, нарушения которых могут стать важным элементом патогенеза опиатной наркомании. Например, есть сведения о вовлечении в механизмы толерантности к опиатами каскада "N-метил-D-аспартатные рецепторы - оксид азота" [44, 49, 50]. Участие N-метил-D-аспартатных рецепторов (NMDA) и глутамата в механизмах толерантности к опиатам доказывается способностью антагонистов названных рецепторов модулировать состояние толерантности [44, 49]. В этом плане проявляют активность и агенты, тропные к участку связывания глицина [49]. Места рецептирования глицина в пределах NMDA-рецепторов играют важную роль в регуляции указанных нервных окончаний [51, 52]. Модуляция нейрональной активности посредством рецепторов N-метил-D-аспартата предполагает участие протеинкиназы С, оксида азота и системы циклического гуанозинмонофосфата. Есть данные о способности ингибиторов NO-синтазы (фермент, ответственный за синтез NO) и гуанилатциклазы влиять на толерантность к опиатам [49]. Одной из структур головного мозга, вовлеченных в развитие толерантности и физической зависимости, считается голубое пятно (locus coeruleus). Предполагается, что в голубом пятне в период формирования абстинентного синдрома активация NMDA нейромедиаторных систем происходит за счет усиления экзоцитоза глутамата, а этот процесс, в свою очередь, медиируется каппа-опиоидными рецепторами [50]. В этой связи поиск фармакологических средств, влияющих на процессы высвобождения глутамата, представляется весьма перспективным для современной наркологии [9, 53]. Менее определенно можно говорить о роли NMDA-рецепторов в модуляции систем вознаграждения при опиатной наркомании. Так, в nucleus accumbens эта система вовлечена в регуляцию поведения самовведения кокаина, но не героина [54]. Важным элементом патогенеза опиатной наркомании в последние годы рассматривают так называемую антиопиоидную систему, включающую неопиоидные пептиды FF, орфанин (ноцицептин) и Tyr-W-MIF-1 [34, 35, 55]. Но о практическом применении агентов, модулирующих эти системы, говорить, по-видимому, преждевременно. III. Изменения генома при опиатной наркоманииПредставления о мутагенной активности опиатов начали формироваться в 70-х годах 20-го столетия [56, 57]. В последующем были изучены повреждения хромосомного аппарата под действием опиатов на клеточных культурах, в опытах на животных [58, 59, 60]. Если согласиться с точкой зрения, что опиаты обладают мутагенной активностью, то остается вопрос: являются ли они первичными мутагенами, или их следует относить к промоторам, либо они обладают свойствами обоих классов? Не менее важным элементом патогенеза опиатной наркомании является способность аддиктивных веществ инициировать экспрессию определенных участков генома. Структуральная идентификация кластеров генома, вовлеченных в экспрессию при опиатной наркомании, доказана в отношение рецепторов [61, 62, 63, 64, 65] и ферментов метаболизма нейромедиаторов [64, 66, 67, 68, 69], белков, вовлеченных в процессы экзоцитоза нейротрансмиттеров [70, 71, 72], а также ионных каналов [73, 74]. Опиаты способны индуцировать изменения экспрессии G-белков [75, 76, 77] и ферментов трансдукторных систем: аденилатциклазы, NO-синтазы, протеинкиназ [78, 79, 80]. Нарушения экспрессии генома выявлены для белков, имеющих отношение к пластическим процессам [81, 82, 83], системам детоксикации [84]. Работы последних лет свидетельствуют, что сдвиги экспрессии мРНК для многих белков зависят от ряда факторов: длительности наркотизации, вида экспериментальных животных, изучаемой структуры головного мозга, длительности абстинентного синдрома и пр. [61, 64, 67]. В качестве примера можно привести исследование японских авторов, посвященное оценке экспрессии гена препроэнкефалина в условиях наркотизации крыс морфином [69]. Содержание мРНК препроэнкефалина определяли в околопроводном сером веществе (ОСВ) - структуре головного мозга, вовлеченной в формирование физической зависимости при опиатной наркомании. Крысы Спрэйг-Доули 11 дней получали морфин в нарастающей дозе (40-160 мг/кг). Через 2 часа с момента последней инъекции наркотика вводился налоксон (5 мг/кг), что индуцировало синдром отмены (precipitated withdrawal). В ростральном ОСВ изучаемый показатель достоверно понижался через 4 часа после начала абстинентного синдрома, но к 12 часу он возвращался к исходному значению. В каудальном отделе околопроводного серого вещества наблюдалась иная картина: содержание мРНК было достоверно повышено в промежутке 4 часа - 2 дня абстиненции (общий срок наблюдения - 4 дня). У второй группы животных концентрацию мРНК оценивали на протяжении 10 дней на фоне спонтанно развивающегося абстинентного синдрома. Оказалось, что в таких условиях эксперимента в ростральном ОСВ значения показателя не отличались от нормы, а в каудальном отделе структуры уровень мРНК препроэнкефалина был достоверно повышен с первых суток по третьи. По мнению авторов, выявленные изменения носят компенсаторный характер в ответ на ослабление энкефалинергической нейротрансмиссии в период наркотизации и абстинентного синдрома. При этом отмечается, что достаточно компактная структура околопроводное серое вещество в нейрохимическом и физиологическом отношениях является гетерогенной [69]. Не ясно, носят ли изменения экспрессии генома транзиторный характер и всегда ли они коррелируют с нарушениями функционального состояния соответствующих кластеров ДНК? Не всегда понятно: нарушения какого уровня передачи генетической информации являются решающими? Показано, например, что сдвиги экспрессии мРНК могут инициироваться как на уровне транскрипции, так и посредством посттранскрипционных механизмов [66, 81]. Известно, что при хронической наркотизации морфином возрастает активность тирозингидроксилазы - лимитирующего фермента синтеза катехоламинов. Эти изменения обнаружены в голубом пятне и в вентральной области покрышки - важных структурах системы вознаграждения. Причем, если в голубом пятне выявленный феномен коррелирует с изменениям экспрессии на уровне транскрипции, то в вентральной области покрышки его основой служат посттранскрипционные механизмы [66]. С другой стороны, морфин, вводимый в желудочки мозга, подавляет экспрессию орнитиндекарбоксилазы печени шестидневных крыс по обоим механизмам [81]. По видимому, в механизмы модуляции генома при опиатной наркомании вовлечены системы вторичных посредников. Так, циклический аденозинмонофосфат влияет на ДНК через специальный белок ssCRE-BP (single-stranded cyclic AMP response element-binding protein), на который, в свою очередь, действуют другие белки-регуляторы. Длительная наркотизация опиатами изменяет активность названной системы, свидетельствуя тем самым о важной роли таких сдвигов в механизмах толерантности и зависимости [85, 86, 87]. Нейрохимиками Университета Осака (Япония) ядерный фактор ssCRE-BP охарактеризован как полипептид с молекулярной массой 110-150 kDa. Он включает два домена, содержащих значительное число остатков глицина и глутамина. Выделена также мРНК, кодирующая изученный белок [87, 88]. Предполагается, что при опиатной наркомании и другие трансдукторные системы способны инициировать изменения функционального состояния генома посредством фосфорилирования белков-регуляторов. Если исходить из классических представлений о структурно-метаболических комплексах клетки [89], можно утверждать, что при опиатной наркомании имеет место модуляция генов основных звеньев данной системы. Как следствие таких нарушений многие авторы рассматривают формирование толерантности, пристрастия и зависимости [61, 65, 68, 69, 82]. Так, нарушения экспрессии мРНК препроэнкефалина в ростро-каудальном околопроводном сером веществе коррелируют с симптомами синдрома отмены [69]. Дисфункция системы мРНК - мускариновые рецепторы М2 в области сосудодвигательного центра крыс на пике абстиненции могут иметь отношение к формированию вегетативных нарушений при отмене опиатов [90]. Изменения экспрессии гена, кодирующего синтез ингибитора связывания (DBI, эндогенный лиганд бензодиазепиновых рецепторов), по мнению М. Катсуры и др. [91], в определенной степени объясняют явления тревожности, агрессивности, психотические сдвиги, наблюдающиеся в период абстинентного синдрома. В механизмах толерантности и зависимости задействованы нарушения экспрессии мРНК, кодирующих синтез различных форм гуаниннуклеотидсвязывающих белков и протеинкиназ [80]. Сложнее экстраполировать данные о сдвигах содержания мРНК в плане формирования феномена пристрастия. Основная проблема - временные несоответствия между изменениями экспрессии генома и состоянием пристрастия - последний по длительности несопоставимо больше. Вероятно, ситуация будет проясняться в процессе накопления новой информации об изменениях функционального состояния генома, более глубокого изучения нарушений транскрипции, процессинга, сплайсинга и трансляции при опиатной наркомании. В формировании психической зависимости, несомненно, задействованы и механизмы долговременной памяти, реализующиеся на уровне генома. Это направление исследований представляется перспективным в плане реабилитации больных наркоманией. Таким образом, нейрохимические исследования, посвященные патогенезу опиатной наркомании, являются бурно прогрессирующей областью современной медицины. Накапливающиеся научные факты расширяют представления о механизмах формирования толерантности, абстинентного синдрома, пристрастия. Это позволяет с определенным оптимизмом смотреть на перспективы лечения и реабилитации больных наркоманией.
Источник: Ваша оценка:
|
Пользователей онлайн: 0. Всего гостей: 1 Поисковики: нет. 
Приветствую вас на своем сайте, здесь вы можете найти много полезной информации (или что-то типа того) САЗОНОВ Вячеслав Фёдорович доцент кафедры биологии Рязанского государственного университета имени С.А. Есенина, кандидат биологических наук. Преподаватель вуза с 1978 года... РекламаПоискПритча наудачу:На сайте введена регистрация через социальные сети, если вы хотите оставлять комментарии без потверждения, пожалуйста, воспользуйтесь именно этим типом аутентификации. Если у вас уже есть аккаунт на сайте, вы можете привязать его к любой социальной сети? зайдя в настройки вашего аккаунта("Мои учётные данные") ниже и воспользовавшись вкладкой "Подключение к социальным сетям". После того, как вы зайдёте при помощи аккаунта в социальной сети, ваши возможности на сайте возрастут. Поддержка сайтаДанный сайт "Кинезиолог" (kineziolog.su) - продукт чисто человеческой "мозговой работы" (термин предложен автором сайта по аналогии с термином "ручная работа" или «handmade»): "мозговая работа" = «brainmade» © 2026 Сазонов В.Ф. © 2026 kineziolog.su). Это означает, что данный сайт создан и поддерживается за счёт чисто человеческой умственной деятельности, без использования нейросетей и/или искусственного интеллекта. Вы можете поддержать сайт своим добрым словом, лайком и/или полноценной ссылкой на его материалы! Это очень поможет. В ответ на вашу поддержку IT-специалисты, следящие за сайтом день и ночь, хотя бы удовлетворённо улыбнутся. ) -26-07-26-05-37 |
||||||||||||||||||||
 ВКонтакте
ВКонтакте Facebook
Facebook Twitter
Twitter